转自:http://blog.sciencenet.cn/home.php?mod=space&uid=635619&do=blog&id=884213
//今天看到一篇非常好的讲解RNA-seq的文章,mark一下。
1.基本步骤
RNAseq分析大致分下面几个步骤:
①首先要把测到的序列map到基因组上,
②然后根据map到的区段对细胞构建转录本,
③然后比较几种细胞的转录本并且合并,
④最后衡量差异和可变剪切和其他的分析。
2.mapping
可以使用哈希的方法比对,但是由于基因组重复序列太高,效率很低;
所以有了Burrows-Wheeler变换,BWA,Bowtie 和SOAP2都是用它。
Burrows-Wheeler变换是一种文本压缩算法,对于一个精确的序列查找,最多在给定序列的长度的次数里就能找到匹配。
重要问题***:
因为一条RNA不一定是一个外显子表达出来的,也有可能是几个外显子结合在了一起,原来基因里的内含子被空了出来,这些内含子的长度从五十到十万个碱基不等;
如果直接用DNAseq的方法的话去在基因组里寻找,有些正好在两个exon连接处的序列就会有错配,而且有些在进化过程中遗漏下来的假基因是没有intron的,这样就导致有些序列会被map到假基因上去,使假基因的表达变得很高,所以,传统的bwa和bowtie在RNAseq里都不是最好的选择。
3.构建转录本
Mapping完了以后,cufflinks就可以把map到基因组里的序列组装成一个转录组了,这个转录组理论上包含了所有当时细胞里的所有mRNA,组装好的转录组包含了可能的剪切信息和所有转录的表达量,这个表达量是根据map到基因组的序列的总数和每个转录片断的长度进行归一化的,听起来比较难懂,它是对于在转录片断里的每一千个碱基对,在每一百万个成功map的序列中,map在这一千个碱基对上的序列的比例,fragments per kilobase of transcript per million mapped fragments (FKPM)。
计算公式:
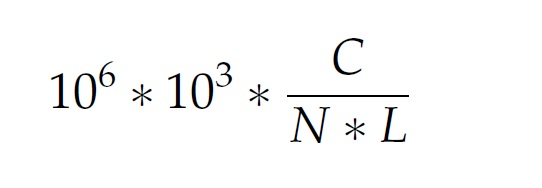
在公式里,C代表的是map在这一千个碱基对上的序列的个数,N是所有成功map的序列的个数,L是转录片断的长度。